


Gene Editing Drugs from the EMA Review Perspective
By Yujiao Zhang
June 25, 2024
Featured Products





Casgevy, the world's first drug based on CRISPR/Cas9 gene-editing technology, was approved for market release at the end of 2023, marking a significant milestone for gene-editing drugs. While the speed at which gene-editing technology is being developed for public benefit is exhilarating, research into gene-editing medicinal products is still in its early stages, and the current regulatory framework offers limited scientific guidance. In April 2024, authors from the European Medicines Agency (EMA) published an article in Nature Reviews titled "Genome-editing medicinal products: the EMA perspective", analyzing the expectations and considerations of regulatory agencies for such drugs from the perspective of the EMA. The viewpoints presented are highly valuable for our attention and reference, and some content is shared below.

Figure 1. Characteristics of Nine Gene Editing Products by EMA [1]
Quality-Related Challenges
Focus areas involve the characterization and control of gene-editing drugs, design of starting materials, definition of active substances, excipients and impurities, potency testing, and comparability during the development process. These aspects need early attention during the development of gene-editing drugs. Like other complex Advanced Therapy Medicinal Products (ATMPs), the quality control strategy for gene-editing drugs should be based on risk assessment, maintaining strict manufacturing and control standards while ensuring flexibility.
Characterization and control of gene-editing drugs should focus on starting materials, active substances, identification, purity, and potency definitions. The definitions of starting materials, active substances, and excipients should follow existing guidelines (including GMP aspects). Gene-editing tools used in cells modified in vivo or in vitro are considered important active substances (refer to the EMA website for Quality: active substance). If new scientific advancements are not reflected in the guidelines, sponsors can seek scientific advice (SA) from the EMA. Vectors of gene-editing tools are classified as excipients, and these vectors should typically meet the requirements for novel excipients.
Product and process-related impurities can affect clinical safety and should be fully studied and characterized. For example, the sequence design and purity of gRNA are directly related to off-target risks. For cells modified in vitro, gene-editing tools are considered process-related impurities that need to be removed from the final product. Ensuring these impurities are removed or controlled to safe levels is necessary and important.
Potency testing should relate to the expected biological function and consider on-target editing efficiency. The amount of on-target editing should be identified and quantified. Suitable predictive bioactivity potency assays should be established early in product development to support comparability in clinical and commercial processes, demonstrating that the products used in clinical trials can represent the final commercial products.
Non-Clinical Challenges
Beyond quality, the main challenges in non-clinical development involve off-target toxicity evaluation, selection of animal models, biodistribution assessment, and developmental and reproductive toxicity studies.
Off-target toxicity is the most important safety concern for gene-editing drugs. Although there is currently no gold standard for off-target evaluation, appropriate and adequate off-target assessments are crucial to minimize safety risks. Developers need to detail the methodology for identifying off-target sites, and the methods used must assess potential biological consequences and related safety issues. From a safety perspective, even low-frequency off-target events are concerning, as they can lead to serious consequences, such as mutagenesis. Off-target toxicity should be evaluated using the final product, and non-clinical safety and toxicity studies should use representative clinical doses, administration modes, and regimens.
Animal models should be selected based on their relevance to clinical settings, and developers should demonstrate their advantages and limitations, including anatomical and functional comparability of target tissues. Follow-up periods should be sufficient to document long-term efficacy, local tolerance, and systemic toxicity.
Biodistribution studies should be conducted in appropriate in vivo models to determine the product's biodistribution at the administration site, identify potential off-target toxicity, and study its pharmacokinetics. Biodistribution analysis must include reproductive organs and potential germline transmission.
Clinical Challenges
Gene-editing drugs mainly target rare diseases, with small patient populations but significant individual variability. It is recommended to seek scientific advice (SA) from the EMA early in the clinical trial phase to discuss patient populations, doses, endpoints, efficacy, and safety follow-ups. The duration and long-term follow-up of pivotal trials are crucial. For gene-editing drugs, a 15-year follow-up is considered appropriate, and the EMA encourages developers to discuss and determine the details of safety and efficacy studies with regulatory agencies.
Quality and non-clinical study data are closely related to the safety and efficacy of clinical trials. Therefore, developers are encouraged to seek scientific advice (SA) early in the product development phase, allowing regulatory agencies to better understand potential challenges that may impact marketing application clinical requirements.
Conclusion
The market approval of Casgevy is a significant milestone for gene-editing drugs, and the attitudes and recommendations from regulators are valuable references for other gene-editing drugs. As of now, Casgevy has been approved for market release in the EU, the USA, Saudi Arabia, and other regions.
High Standard Gene Editing Enzymes from KACTUS
KACTUS is a protein raw material company driven by independent innovation, focusing on serving the development and production of biopharmaceuticals. Since its establishment in 2018, KACTUS has accumulated rich experience in protein research and production through its unique SAMSTM protein research platform based on structural design.
KACTUS brings together a technical and professional team of nearly 200 people, creating a research and production center and a tens of thousands of square meters GMP-grade protein enzyme industrialization base, providing strong support for global drug research and development.
KACTUS provides various GMP-grade raw proteins and enzymes to meet the development needs of cell and gene therapy drugs. The lineup includes multiple types of gene editing enzymes such as GMP-grade Cas9 and the independently developed GMP-grade base editor AccuBaseTM (by Base Therapeutics). The GMP-grade Cas9 has completed FDA DMF filing and has provided strong support for multiple IND submissions.
Below is a list of related products (click on the product code for details):
|
Catalog Number |
Product Information |
|
Cas9 Nuclease 【100μg/1mg】 |
|
|
Cas9 Nuclease, GMP grade 【3mg】 |
|
|
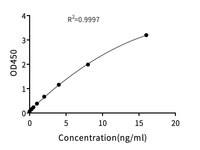
Cas9 (CRISPR Associated Protein 9) ELISA Kit 【96T】 |
|
|
SpCas9 D10A Nickase 【100μg/1mg】 |
|
|
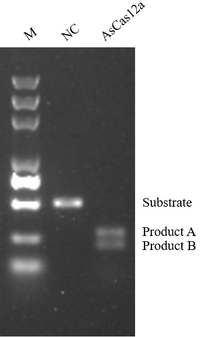
AsCas12a Nuclease 【100μg/1mg】 |
|
|
BS-EP1 (AccuBaseTM) 【100μg/200μg/500μg/1mg】 |
|
|
BS-EP1 (AccuBaseTM), GMP grade 【1mg】 |
References
[1] Tavridou A, Rogers D, Farinelli G, Gravanis I, Jekerle V. Genome-editing medicinal products: the EMA perspective. Nat Rev Drug Discov. 2024 Apr;23(4):242-243. doi: 10.1038/d41573-024-00050-2.



60 Hickory Drive
Waltham, MA 02451
United States